Ako bolo už viac krát spomenuté v kvapalnej fáze sú informácie o dIS pre ich efektívne spriemerňovanie na nulu nedostupné. Napriek tomu pretrvávala v NMR komunite neustála snaha o využitie ich veľkého potenciálu aj pre štruktúrnu analýzu kvapalných vzoriek. Jednou z hlavných motivácií je fakt, že zatiaľčo štandardné NMR parametre (chemický posun, multiplicita signálov, veľkosť J, …) poskytujú iba lokálne štruktúrne informácie o okolí spinov na ktoré sa vzťahujú, DD interakcie môžu poskytovať aj štruktúrne informácie „ďalekého dosahu“ (long-range). V kvapalnej fáze je to nový typ štruktúrnej informácie
Vedelo sa, že jedinou možnosťou ako to dosiahnuť je čiastočná orientácia molekúl v kvapalnej fáze. Zorientovanie vzorky sa môže dosiahnuť rozličným spôsobom, napr. jej rozpustením v kvapalno-kryštalickom prostredí [9], pridaním magneticky anizotropnej látky, ktorá sa v smere B0 čiastočne orientuje alebo umiestnením v priestorovo anizotropnej matrici (mechanický upravene polymérne gély).
Stupeň zorientovania molekúl x je možno zjednodušené vyjadriť ako zlomok úplne zorientovaných molekúl z celkového počtu molekúl prítomných vo vzorke. Molekuly sú v rýchlej výmene medzi zorientovaným a nezorientovaným stavom (mólový zlomok = 1- x, dIS spriemernená na nulu). Táto rýchla výmena spôsobí spriemernenie dIS v celej vzorke na tzv. redukovanú hodnotu dISr:
dISr= x.dIS + (1-x).0= x.dIS
V odbornej literatúre sa na označenie dISr zaužívalo označenie RDC (Reduced Dipolar Coupling). Okrem redukcie dIS sa v kvapalnej fáze v dôsledku tepelného pohybu medzimolekulové DD interakcie sa prakticky vynulujú (čo znamená zmenšenie počtu medzi sebou interagujúcich spinov) a predlží sa relaxačný čas T2 (zúženie spektrálnych čiar). To všetko spôsobuje zjednodušenie spektier oproti ekvivalentným spektrám v tuhej fáze.
Ako bolo spomenuté vyššie, čiastočné zorientovanie molekúl v kvapalnej fáze možno dosiahnuť rôznymi spôsobmi, no ukázalo sa, že pre praktické aplikácie je potrebné aby zorientovanie molekúl bolo relatívne malé a kontrolovateľné.
Molekuly s paramagnetickým centrom alebo silnou magnetickou anizotropiou (veľké asymetrické molekuly ako napr. polynukleotidy,..) sa sami čiastočne orientujú v silnom magnetickom poli B0. Pre štandardné diamagnetické molekuly je však stupeň orientácie v dostupných magnetických poliach zanedbateľný, a preto sa tento najjednoduchší spôsob zorientovania molekúl dá prakticky využiť len veľmi obmedzené.
Na zorientovanie molekúl možno využiť vhodné kvapalno-kryštalické média. Komerčne sú dostupné rôzne média. Potrebné je vybrať také, v ktorom sa analyzovaná látka dostatočne rozpúšťa a médium si zachováva anizotropný charakter aj pri malej koncentrácií mezogénov (molekúl vytvárajúcich kvapalno-kryštalickú štruktúru).
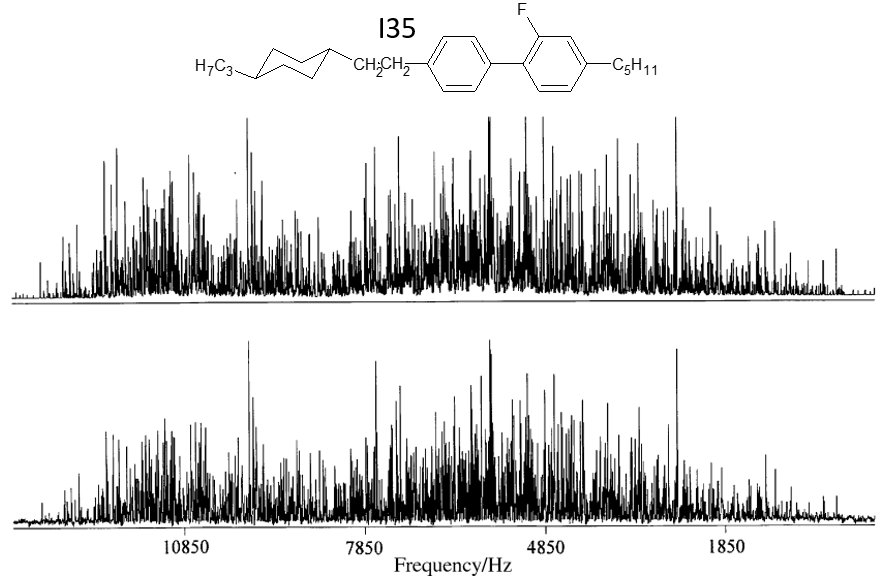
Stupeň usporiadania molekúl analytu v médiách, ktoré boli v 60tich až 90tich rokoch minulého storočia dostupné bol relatívne vysoký (x ~ 0.1). Takýto stupeň usporiadania molekúl znamená efektívnu redukciu hodnôt dIS na desatinu statických hodnôt (pozri tabuľku a obr.56 ), čo ale stále pre blízko vzdialené spiny (priamo viazané: C-H; N-H, H-C-H,..) znamená, že redukované hodnoty dISr budú na úrovni niekoľko stoviek až tisíc Hz a taktiež to znamená existenciu veľkého počtu nezanedbateľných interakcií medzi vzdialenejšími spinmi. Veľký počet významných dISr interakcií medzi spinmi spôsobuje pri väčších spinových systémov prílišnú komplikovanosť spektier. Na ilustráciu je na obr.57 uvedené spektrum etyl benzénu v komerčnom nematickom rozpúšťadle I35 [10].
Analýza takýchto spektier je však veľmi náročná. Používajú pri nej rôzne stratégie, včítane zjednodušovania spektier selektívnou náhradou vodíka v molekule skúmanej látky za deutérium a využitia sofistikovaných výpočtových metód [11]. Pri úspešnej analýze sa získajú cenné štruktúrne informácie o študovanej látke (presná geometrická štruktúra, zastúpenie konformérov,..). Je však zrejme, že táto metodika nebude mať nikdy rutinný charakter.
Z praktického hľadiska sú najdôležitejšie slabo orientujúce média, ktoré umožňujú zorientovanie molekúl analytu na úroveň cca 0,1% [12-14]. Takéto média sa objavili až v 90tich rokov minulého storočia. Sú rôzneho charakteru. Okrem špeciálnych kvapalno-kryštalických médií patria sem aj vodné roztoky rôznych nadmolekulových častíc s veľkou magnetickou anizotropiou (bicely, fágy, vírusy,..) a polymérne gély s anizotropnými kavitami.
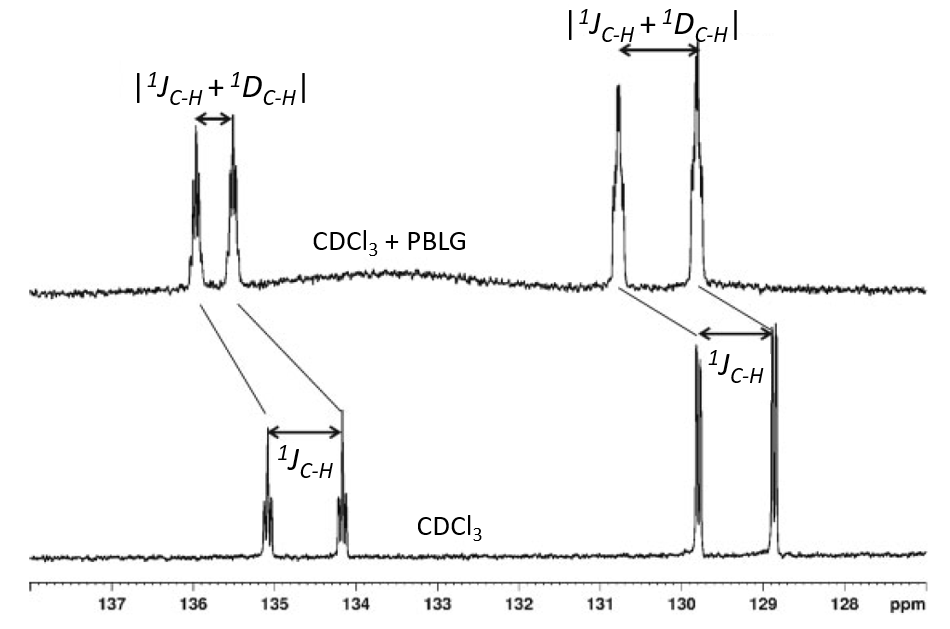
Pri takomto zorientovaní molekúl sú dISr najväčších DD interakcií na úrovní ~10 Hz. Interakcie medzi vzdialenejšími spinmi sa stávajú zanedbateľnými. Výsledné spektrá sú preto veľmi podobné spektrám izotropných vzoriek a sú relatívne ľahko analyzovateľné. DD-interakcia sa prejaví ako príspevok k existujúcej J-interakcií medzi dvojicou spinov. V spektre sa teda pozoruje celková interakcia (TIS) medzi dvojicou spinov I a S ako súčet ich J- a DD interakcie: TIS = JIS + DISr. DISr sa dá určiť z dvoch experimentov ako rozdiel celkovej interakcie TIS v orientovanom a JIS v izotropnom médiu.
Na nasledujúcom obrázku je ilustrovaný efekt slabej orientácie molekúl na vzhľad a charakter 13C NMR spektra nitrobenzénu v chloroforme [13].
Štruktúrna analýza pomocou RDC sa v súčasnosti naviac využíva pri riešení štruktúry izotopovo ( 15N alebo aj 13C) obohatených proteínov. Analýza je zameraná najmä na RDC interakcie medzi priamo viazanými spinmi, hlavne na jednoväzbovú interakciu 1H-15N. Veľkou výhodou, že vzdialenosť medzi spinmi (rN-H ~ 1.0 Å) ako aj veľkosť 1JN-H (JN-H ~ 93 Hz) sú známe a s pohybom molekúl sa menia veľmi málo. Pre 15N izotopovo obohatené je určovanie veľkosti a znamienka v súčasnosti viac-menej rutinnou záležitosťou.
Vyhodnotenie štruktúrnych informácií pomocou RDC ak nie je triviálna. Okrem získania dostatočného počtu RDC je potrebné poznať spôsob a stupeň orientácie molekuly (tenzor orientácie (časti) molekuly, „alignment tensor“ [14]). Analýza je pomerne jednoduchá v prípade rigidných molekúl. Takéto molekuly tvoria napr. niektoré proteíny, pre ktoré RDC analýza významne pomáha potvrdiť alebo spresniť ich terciálnu štruktúru. Analyzovateľné sú aj flexibilné molekuly zložené z viacerých rigidných časti (napr. modulárne proteíny). RDC analýza umožňuje spresniť 3D štruktúru jednotlivých modulov ako aj stanoviť stupeň flexibility medzi modulmi. Podobné ako proteíny jednoväzbové RDC sa využívajú aj v prípade izotopovo obohatených fragmentov DNA a RNA molekúl
Zložitejšie je využitie RDC v menších molekulách s prirodzeným izotopovým zastúpením spinov. Analýza je kvôli citlivosti experimentov viac-menej obmedzená na vyhodnocovanie 1H –1H RDC interakcií. Tie majú dobre definovanú vzdialenosť iba pre geminálne vodíky (vodíky viazané na tom istom uhlíku) a vodíky v rigidných cyklických štruktúrach. Napriek tomu záujem o využitie RDC metodík stále rastie. Je to aj preto, že na mnohé zaujímavé štrukturálne otázky môže dať odpoveď jednoduché porovnanie dvoch relevantných RDC, bez nutnosti analýzy celkovej štruktúry molekuly. Napr. odpoveď či sú dve C-H väzby na rigidnej štruktúre orientované paralelné alebo nie. Ak sú paralelné musí mať ich DIS rovnakú hodnotu, inak platí opak. Informácie o možnosti využitia RDC v menších organických molekulách možno nájsť v literatúre [15,16].


